

Full-Length Transcriptome Profiling of Coridius chinensis Mitochondrial Genome Reveals the Transcription of Genes with Ancestral Arrangement in Insects


Abstract
:1. Introduction
2. Materials and Methods
2.1. Full-Length Transcriptome Sequencing
2.2. Data Assessment and Quality Control of RNA Raw Data
2.3. Mitogenome Sequencing
2.4. Mitochondrial Transcriptome Analysis
3. Results
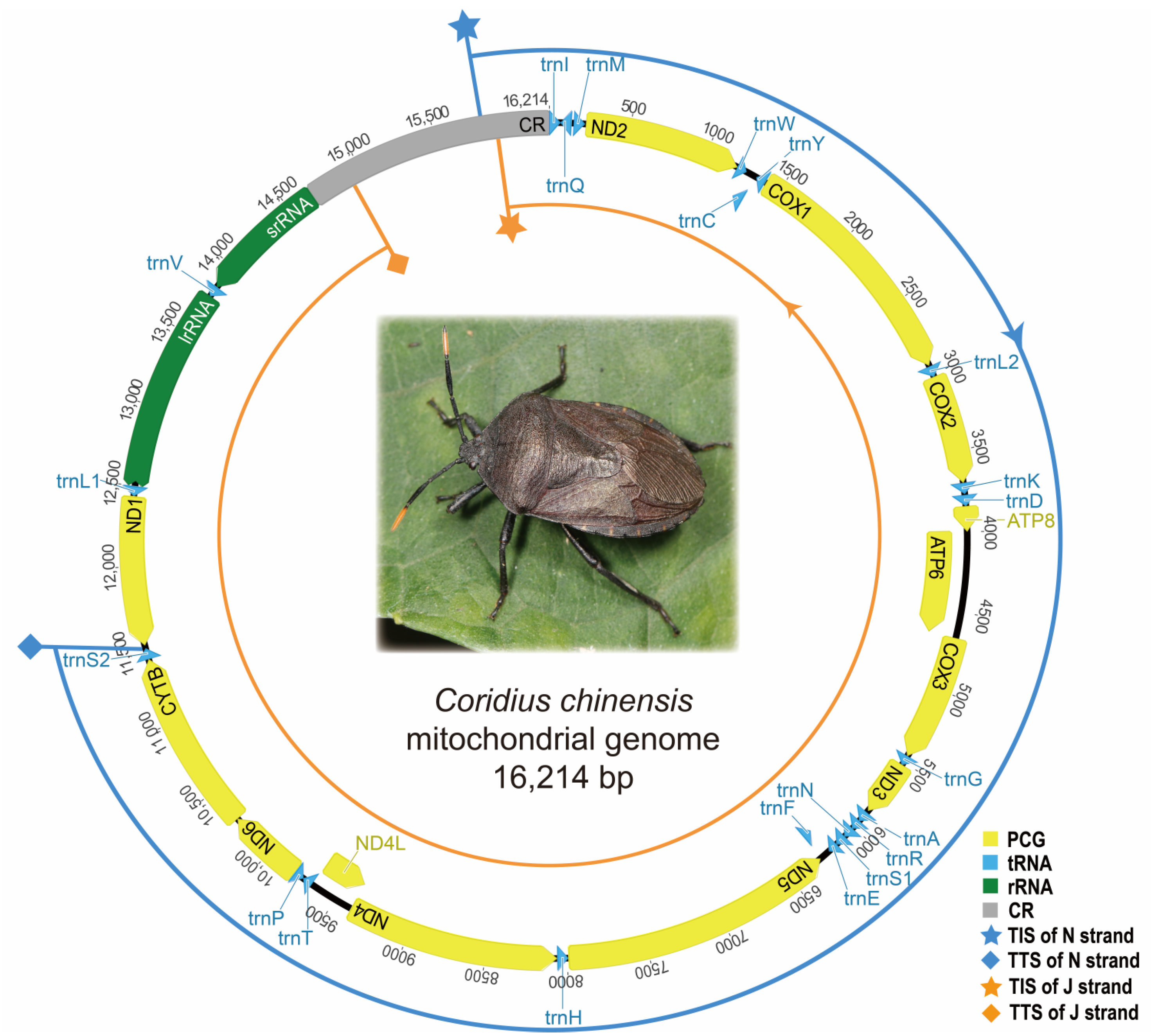
3.1. The Annotation of C. chinensis Mitogenome
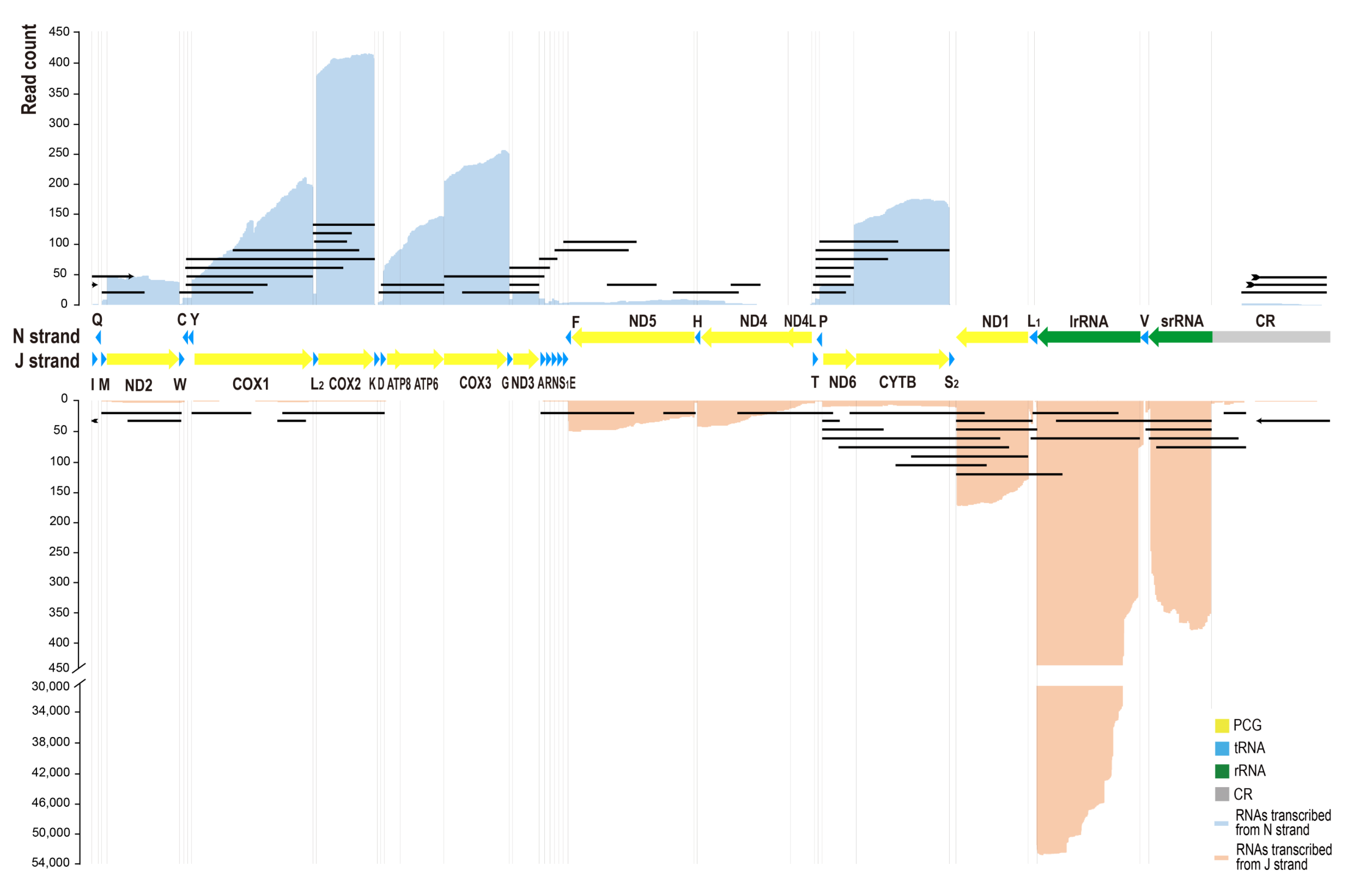
3.2. The Quantitative Transcription Map of C. chinensis Mitogenome
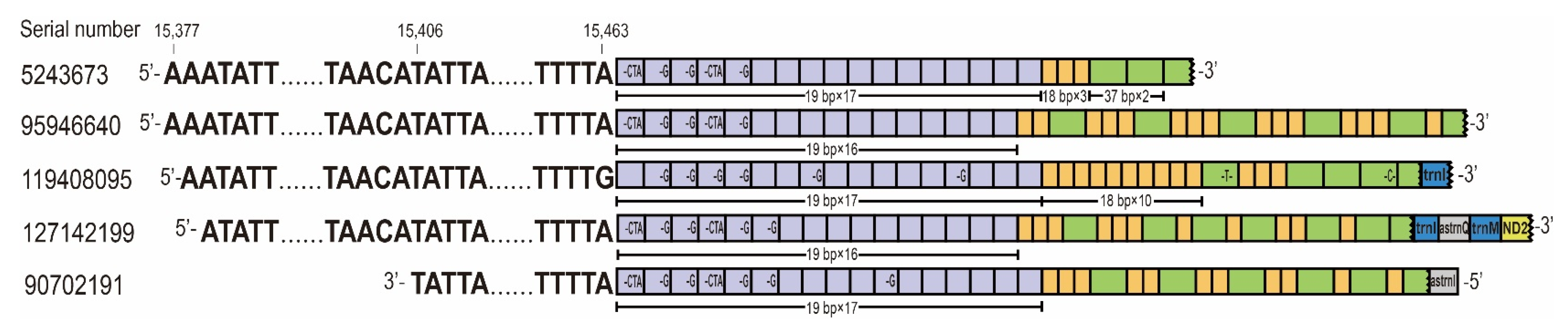
3.3. Detection of lncRNAs from Mitochondrial Control Region
3.4. The Proposed Model of Mitochondrial Transcription
3.5. Transcripts of tRNA and rRNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA transcription and its regulation: An evolutionary perspective. Trends Genet. 2018, 34, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Clary, D.O.; Wolstenholme, D.R. The mitochondrial DNA molecular of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. J. Mol. Evol. 1985, 22, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Berthier, F.; Renaud, M.; Alziari, S.; Durand, R. RNA mapping on Drosophila mitochondrial DNA: Precursors and template strands. Nucleic Acids Res. 1986, 14, 4519–4533. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.B.; Beckenbach, A.T. Characterization of mature mitochondrial transcripts in Drosophila, and the implications for the tRNA punctuation model in arthropods. Gene 2009, 445, 49–57. [Google Scholar] [CrossRef]
- Roberti, M.; Bruni, F.; Polosa, P.L.; Gadaleta, M.N.; Cantatore, P. The Drosophila termination factor DmTTF regulates in vivo mitochondrial transcription. Nucleic Acids Res. 2006, 34, 2109–2116. [Google Scholar] [CrossRef] [Green Version]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Gao, S.; Ren, Y.; Sun, Y.; Wu, Z.; Ruan, J.; He, B.; Zhang, T.; Yu, X.; Tian, X.; Bu, W. PacBio full-length transcriptome profiling of insect mitochondrial gene expression. RNA Biol. 2016, 13, 820–825. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Cheng, Z.; Wang, B.; Yau, T.O.; Chen, Z.; Barker, S.C.; Chen, D.; Bu, W.; Sun, D.; Gao, S. Precise annotation of human, chimpanzee, rhesus macaque and mouse mitochondrial genomes leads to insight into mitochondrial transcription in mammals. RNA Biol. 2020, 17, 395–402. [Google Scholar] [CrossRef]
- Gao, S.; Tian, X.; Chang, H.; Sun, Y.; Wu, Z.; Cheng, Z.; Dong, P.; Zhao, Q.; Ruan, J.; Bu, W. Two novel lncRNAs discovered in human mitochondrial DNA using PacBio full-length transcriptome data. Mitochondrion 2018, 38, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Xu, X.; Jin, X.; Yin, H.; Luo, J.; Liu, G.; Zhao, Q.; Chen, Z.; Bu, W.; Gao, S. Using high-resolution annotation of insect mitochondrial DNA to decipher tandem repeats in the control region. RNA Biol. 2019, 16, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xuan, Y.; Liang, G.; Yang, X.; Yu, Z.; Barker, S.C.; Kelava, S.; Bu, W.; Liu, J.; Gao, S. Precise annotation of tick mitochondrial genomes reveals multiple copy number variation of short tandem repeats and one transposon-like element. BMC Genom. 2020, 21, 488. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.X.; Chen, Z.; Li, H.M.; Lou, D.J.; Liu, X.B.; Dai, X.H.; Shi, H.Y. Morphological and biological characteristics of Coridius chinensis (Hemiptera: Pentatomidae). J. Environ. Entomol. 2022, 44, 194–203. [Google Scholar]
- Liu, L.; Li, H.; Song, F.; Song, W.; Dai, X.; Chang, J.; Cai, W. The mitochondrial genome of Coridius chinensis (Hemiptera: Dinidoridae). Zootaxa 2012, 3537, 29–40. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Taft, R.J.; Glazov, E.A.; Cloonan, N.; Simons, C.; Stephen, S.; Faulkner, G.J.; Lassmann, T.; Forrest, A.R.; Grimmond, S.M.; Schroder, K.; et al. Tiny RNAs associated with transcription start sites in animals. Nat. Genet. 2009, 41, 572–578. [Google Scholar] [CrossRef]
- D’Souza, A.R.; Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Margam, V.M.; Coates, B.S.; Hellmich, R.L.; Agunbiade, T.; Seufferheld, M.J.; Sun, W.; Ba, M.N.; Sanon, A.; Binso-Dabire, C.L.; Baoua, I.; et al. Mitochondrial genome sequence and expression profiling for the legume pod borer Maruca vitrata (Lepidoptera: Crambidae). PLoS ONE 2011, 6, e16444. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, J.; Boykin, L.; Zhao, Q.; Li, Q.; Wang, X.; Liu, S. The characteristics and expression profiles of the mitochondrial genome for the Mediterranean species of the Bemisia tabaci complex. BMC Genom. 2013, 14, 401. [Google Scholar] [CrossRef] [Green Version]
- Schürer, H.; Schiffer, S.; Marchfelder, A.; Mörl, M. This is the end: Processing, editing and repair at the tRNA 3’-terminus. Biol. Chem. 2001, 382, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Slomovic, S.; Laufer, D.; Geiger, D.; Schuster, G. Polyadenylation and degradation of human mitochondrial RNA: The prokaryotic past leaves its mark. Mol. Cell Biol. 2005, 25, 6427–6435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokolenko, I.N.; Alexeyev, M.F. Mitochondrial transcription in mammalian cells. Front. Biosci. 2017, 22, 835–853. [Google Scholar]
- Kadaba, S.; Krueger, A.; Trice, T.; Krecic, A.M.; Hinnebusch, A.G.; Anderson, J. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 2004, 18, 1227–1240. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.; Jensen, T.H. The exosome: A multipurpose RNA-decay machine. Trends Biochem. Sci. 2008, 33, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Lunt, D.H.; Whipple, L.E.; Hyman, B.C. Mitochondrial DNA variable number tandem repeats (VNTRs): Utility and problems in molecular ecology. Mol. Ecol. 1998, 7, 1441–1455. [Google Scholar] [CrossRef]
- Mundy, N.I.; Helbig, A.J. Origin and evolution of tandem repeats in the mitochondrial DNA control region of shrikes (Lanius spp.). J. Mol. Evol. 2004, 59, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Solignac, M.; Génermont, J.; Monnerot, M.; Mounolou, J. Genetics of mitochondria in Drosophila: mtDNA inheritance in heteroplasmic strains of D. mauritiana. Mol. Gen. Genet. 1984, 197, 183–188. [Google Scholar] [CrossRef]
- Casane, D.; Dennebouy, N.; de Rochambeau, H.; Mounolou, J.C.; Monnerot, M. Nonneutral evolution of tandem repeats in the mitochondrial DNA control region of lagomorphs. Mol. Biol. Evol. 1997, 14, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome Animals. Comp. Genom. 2000, 1, 133–147. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Gene | Coding Strand | Annotations Using DNA Sequencing | Annotations According to Transcripts | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Position (J Strand as Reference) | Length/bp | Start Codon | Stop Codon | Position (J Strand as Reference) | Length/bp | Start Codon | Stop Codon | ||
| trnI | J | 1-71 | 71 | 1-71 | 71 | ||||
| trnQ | N | 142-73 | 70 | 142-73 | 70 | ||||
| trnM | J | 155-224 | 70 | 155-224 | 70 | ||||
| ND2 | J | 229-1206 | 978 | AUA | UAA | 229-1,204 | 976 | AUA | U |
| trnW | J | 1205-1268 | 64 | 1205-1268 | 64 | ||||
| trnC | N | 1329-1261 | 69 | 1329-1261 | 69 | ||||
| trnY | N | 1402-1337 | 66 | 1402-1337 | 66 | ||||
| COX1 | J | 1407-2942 | 1536 | UUG | UAA | 1407-2942 | 1536 | UUG | UAA |
| trnL2 | J | 2949-3019 | 71 | 2949-3019 | 71 | ||||
| COX2 | J | 3020-3698 | 679 | AUA | U | 3020-3698 | 679 | AUA | U |
| trnK | J | 3699-3770 | 72 | 3699-3770 | 72 | ||||
| trnD | J | 3775-3842 | 68 | 3775-3842 | 68 | ||||
| ATP8 | J | 3844-4002 | 159 | UUG | UAA | 3844-4002 | 159 | UUG | UAA |
| ATP6 | J | 3996-4679 | 684 | AUG | UAA | 3996-4665 | 670 | AUG | U |
| COX3 | J | 4666-5454 | 789 | AUG | UAA | 4666-5453 | 788 | AUG | UA |
| trnG | J | 5454-5518 | 65 | 5454-5518 | 65 | ||||
| ND3 | J | 5516-5872 | 357 | AUA | UAA | 5519-5872 | 354 | AUA | UAA |
| trnA | J | 5881-5949 | 69 | 5881-5949 | 69 | ||||
| trnR | J | 5950-6017 | 68 | 5950-6017 | 68 | ||||
| trnN | J | 6019-6084 | 66 | 6019-6084 | 66 | ||||
| trnS1 | J | 6085-6153 | 69 | 6085-6153 | 69 | ||||
| trnE | J | 6154-6218 | 65 | 6154-6218 | 65 | ||||
| trnF | N | 6282-6217 | 66 | 6282-6217 | 66 | ||||
| ND5 | N | 7990-6290 | 1701 | AUU | UAA | 7990-6290 | 1701 | AUU | UAA |
| trnH | N | 8057-7992 | 66 | 8057-7992 | 66 | ||||
| ND4 | N | 9384-8059 | 1326 | AUG | UAG | 9384-8059 | 1326 | AUG | UAG |
| ND4L | N | 9665-9378 | 288 | UUG | UAA | 9665-9378 | 288 | UUG | UAA |
| trnT | J | 9668-9732 | 65 | 9668-9732 | 65 | ||||
| trnP | N | 9797-9734 | 64 | 9797-9734 | 64 | ||||
| ND6 | J | 9801-10,280 | 480 | AUG | UAA | 9801-10,280 | 480 | AUG | UAA |
| CYTB | J | 10,282-11,418 | 1137 | AUG | UAA | 10,282-11,418 | 1137 | AUG | UAA |
| trnS2 | J | 11,421-11,489 | 69 | 11,421-11,489 | 69 | ||||
| ND1 | N | 12,439-11,510 | 930 | AUG | UAA | 12,433-11,510 | 924 | AUG | UAA |
| trnL1 | N | 12,500-12,434 | 67 | 12,500-12,434 | 67 | ||||
| lrRNA | N | 13,774-12,501 | 1274 | 13,774-12,501 | 1274 | ||||
| trnV | N | 13,841-13,775 | 67 | 13,841-13,775 | 67 | ||||
| srRNA | N | 14,652-13,842 | 811 | 14,661-13,842 | 820 | ||||
| CR | 14,653-16,214 | 1562 | 14,662-16,214 | 1553 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Duan, Y.; Ma, L.; Song, F.; Tian, L.; Cai, W.; Li, H. Full-Length Transcriptome Profiling of Coridius chinensis Mitochondrial Genome Reveals the Transcription of Genes with Ancestral Arrangement in Insects. Genes 2023, 14, 225. https://doi.org/10.3390/genes14010225
Xu S, Duan Y, Ma L, Song F, Tian L, Cai W, Li H. Full-Length Transcriptome Profiling of Coridius chinensis Mitochondrial Genome Reveals the Transcription of Genes with Ancestral Arrangement in Insects. Genes. 2023; 14(1):225. https://doi.org/10.3390/genes14010225
Chicago/Turabian StyleXu, Shiwen, Yuange Duan, Ling Ma, Fan Song, Li Tian, Wanzhi Cai, and Hu Li. 2023. "Full-Length Transcriptome Profiling of Coridius chinensis Mitochondrial Genome Reveals the Transcription of Genes with Ancestral Arrangement in Insects" Genes 14, no. 1: 225. https://doi.org/10.3390/genes14010225