
Identification of Key Differentially Expressed mRNAs, miRNAs, lncRNAs, and circRNAs for Xp11 Translocation Renal Cell Carcinoma (RCC) Based on Whole-Transcriptome Sequencing
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients Identification
2.2. Screening of the DEmRNA, DEmiRNA, DElncRNA, and DEcircRNA
2.3. Enrichment Pathway Analysis of DEmRNA
2.4. Protein-Protein Interaction (PPI) Network and Module Construction of DEmRNA
2.5. Prediction of miRNA Regulation Relationship
2.6. Co-Expression Analysis of DElncRNA-DEmRNA and DEcircRNA-DEmRNA
2.7. KEGG Enrichment Analysis of the DEmiRNA, DElncRNA, and DEcircRNA
2.8. Construction of ceRNA Network
2.9. GEO Data Validation
3. Results
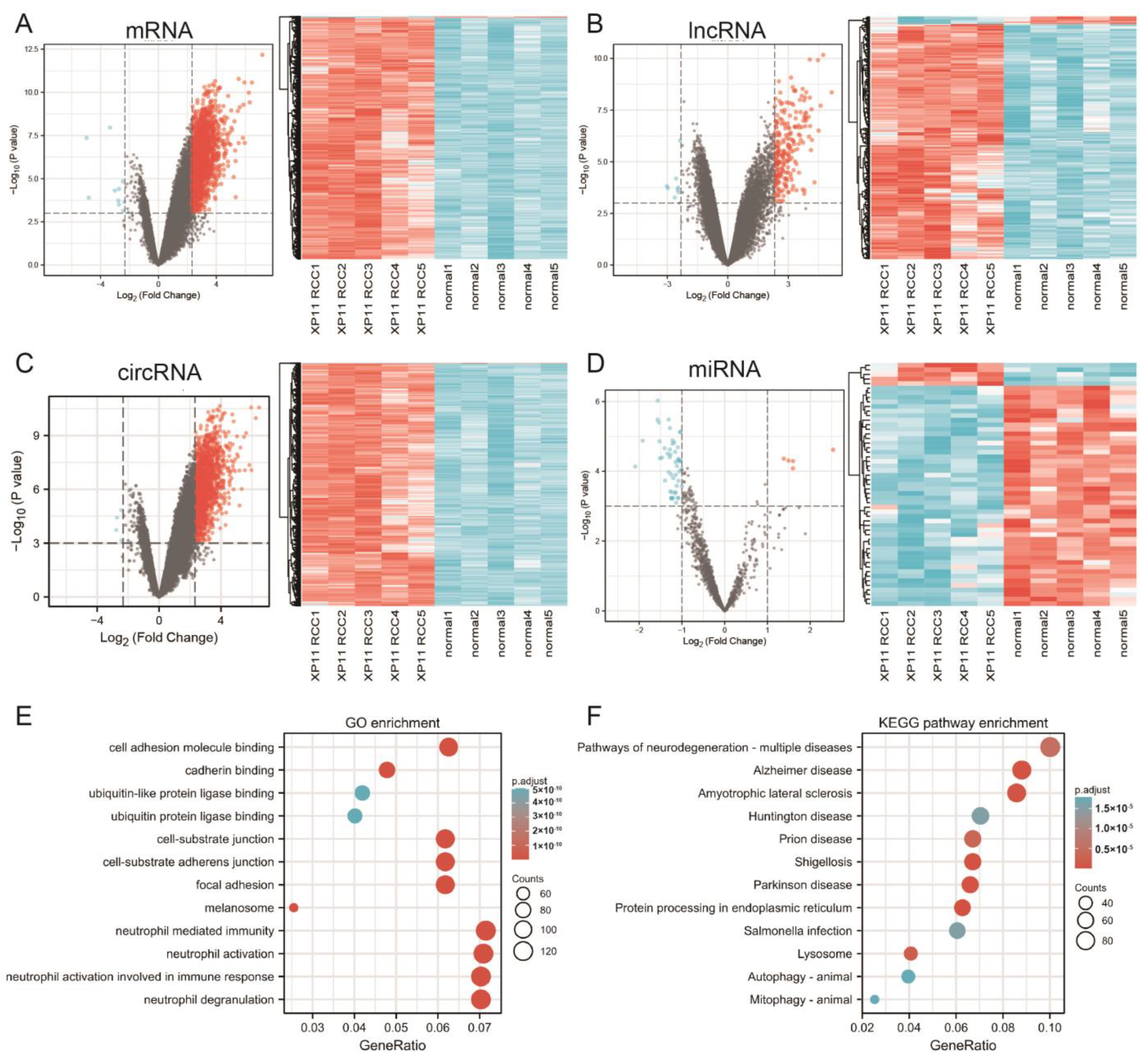
3.1. Differential Expression Analysis
3.2. Functional Enrichment Analysis of DEmRNAs
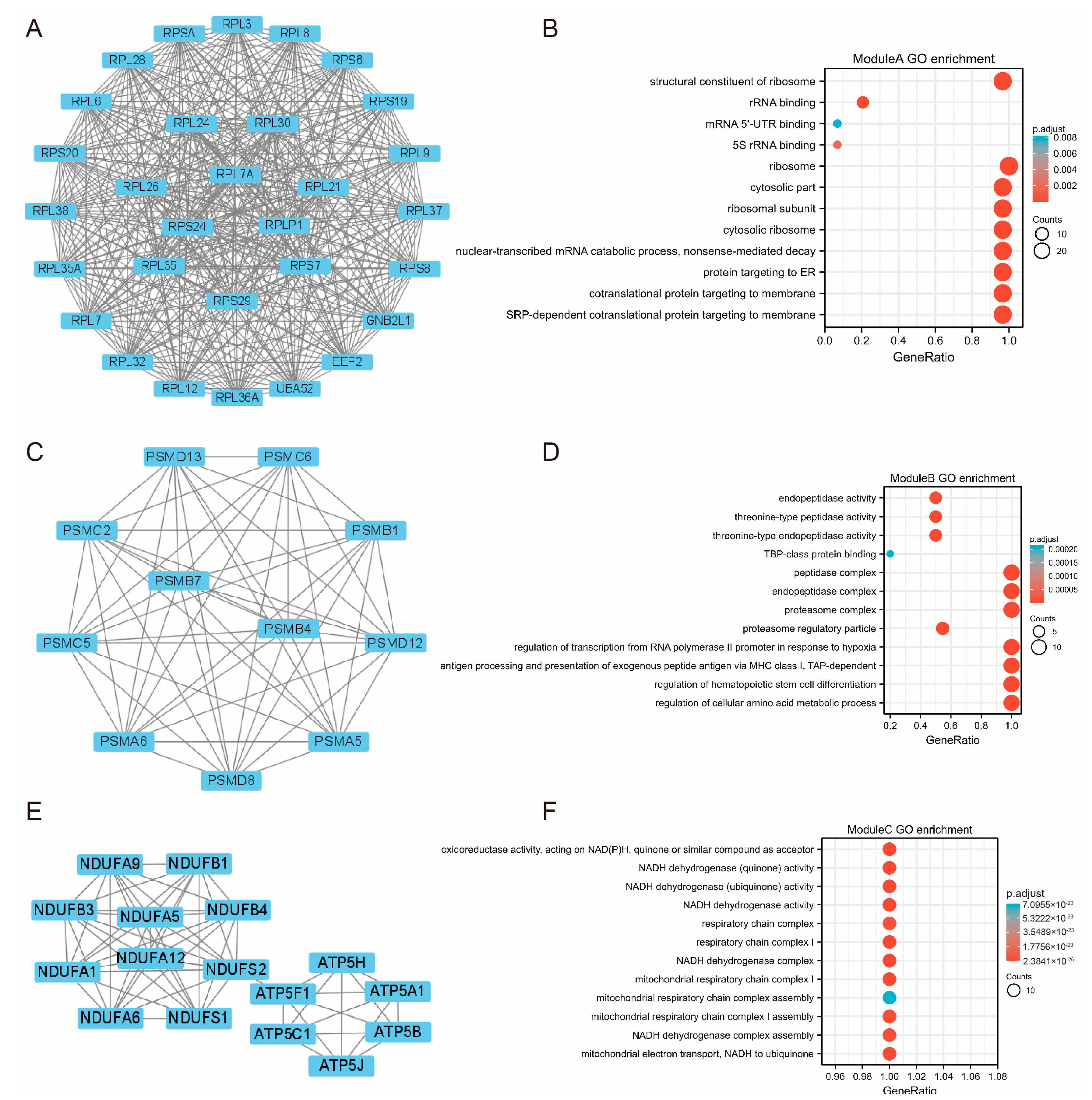
3.3. PPI Network and Module Construction
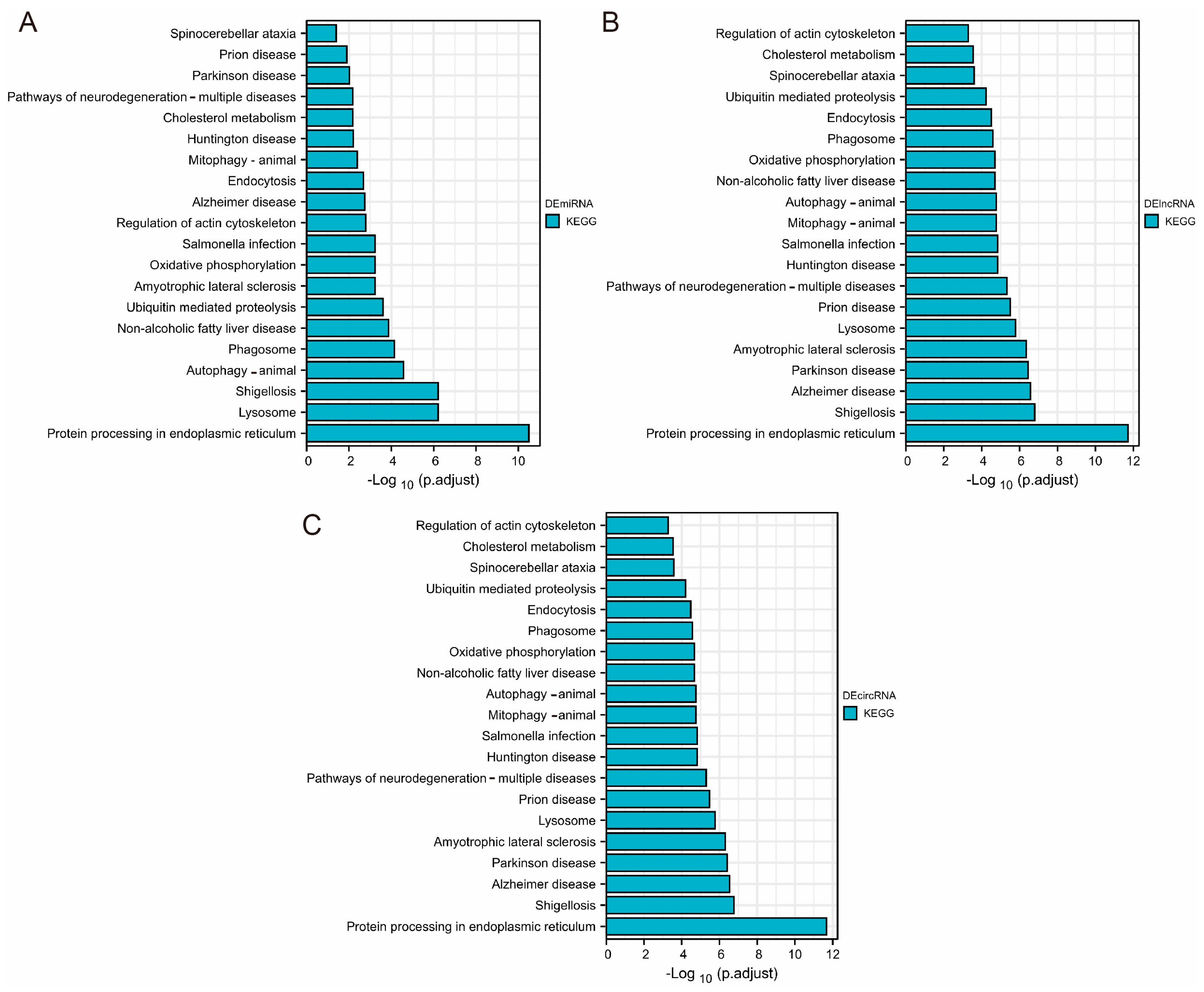
3.4. Enrichment Analysis of DEmiRNA, DElncRNA, and DEcircRNA-Related Target Genes
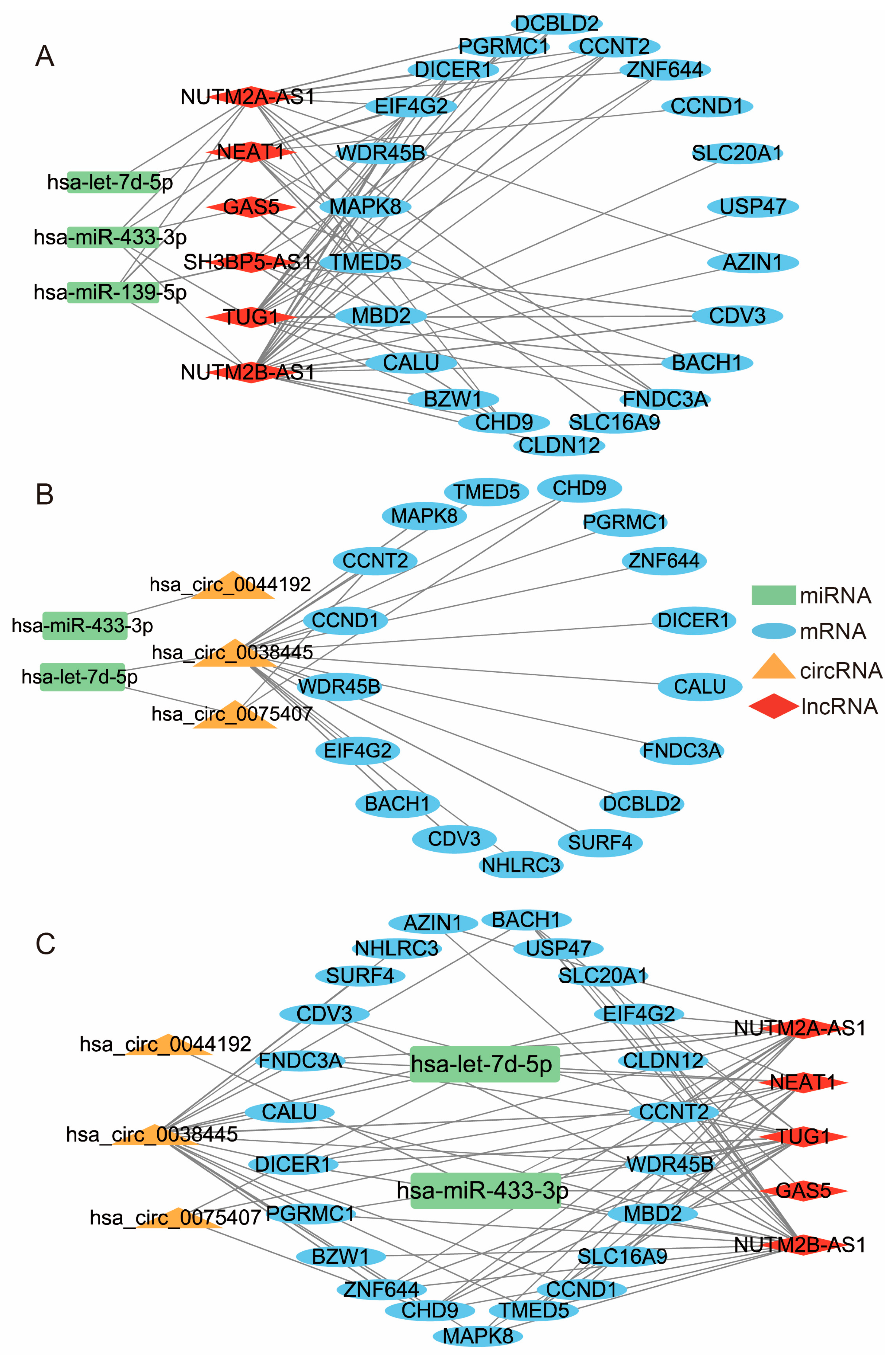
3.5. Construction of ceRNA Network
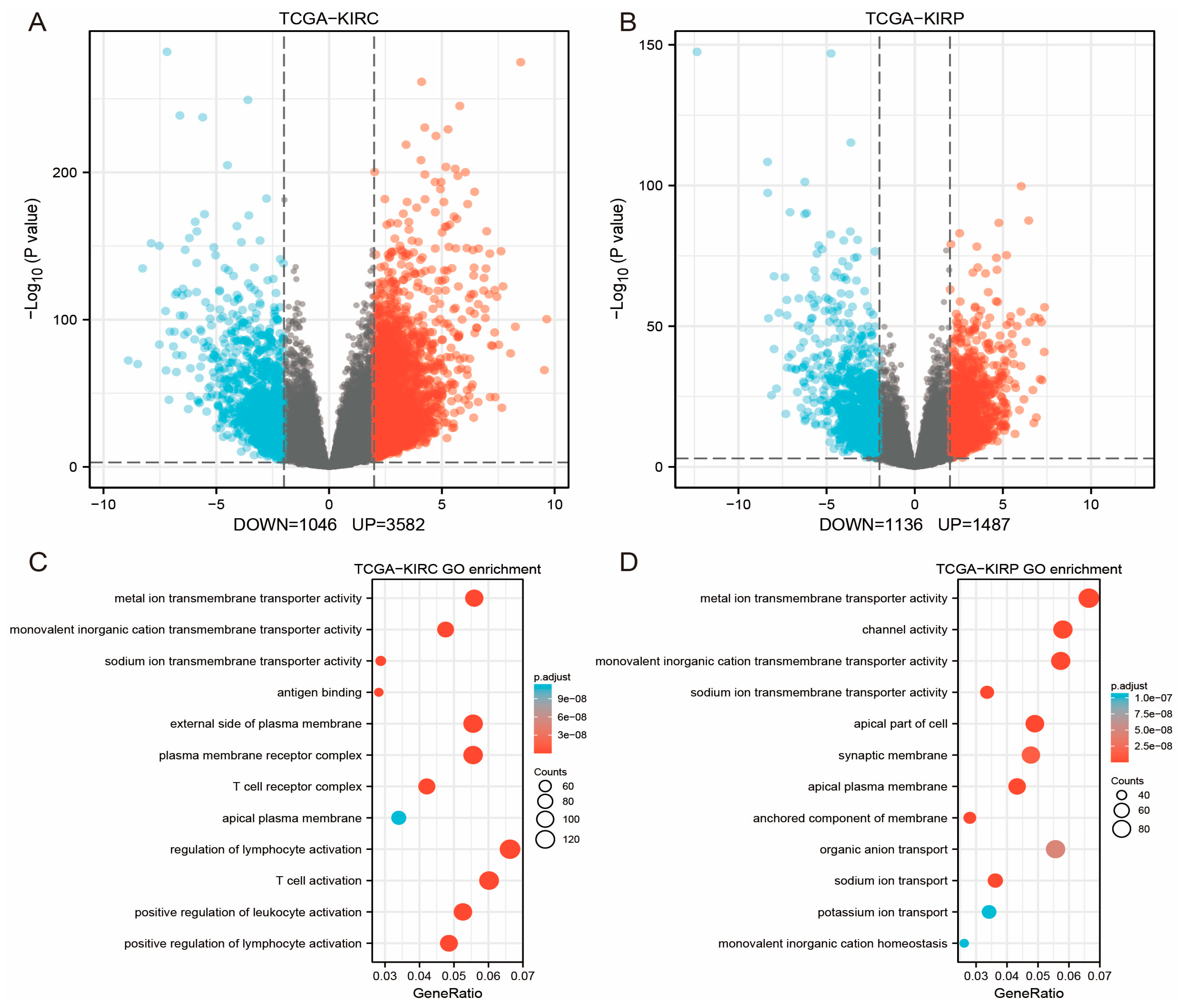
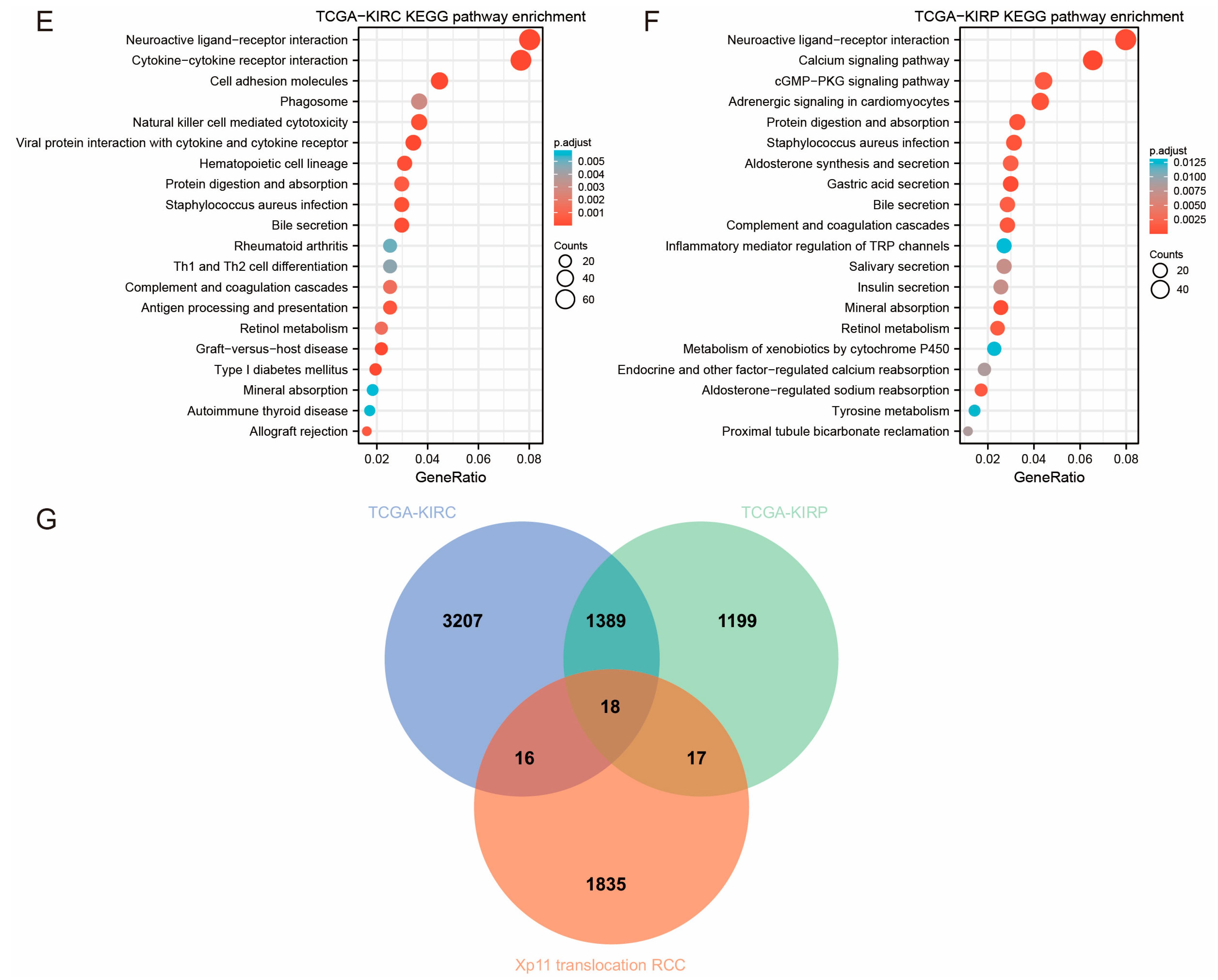
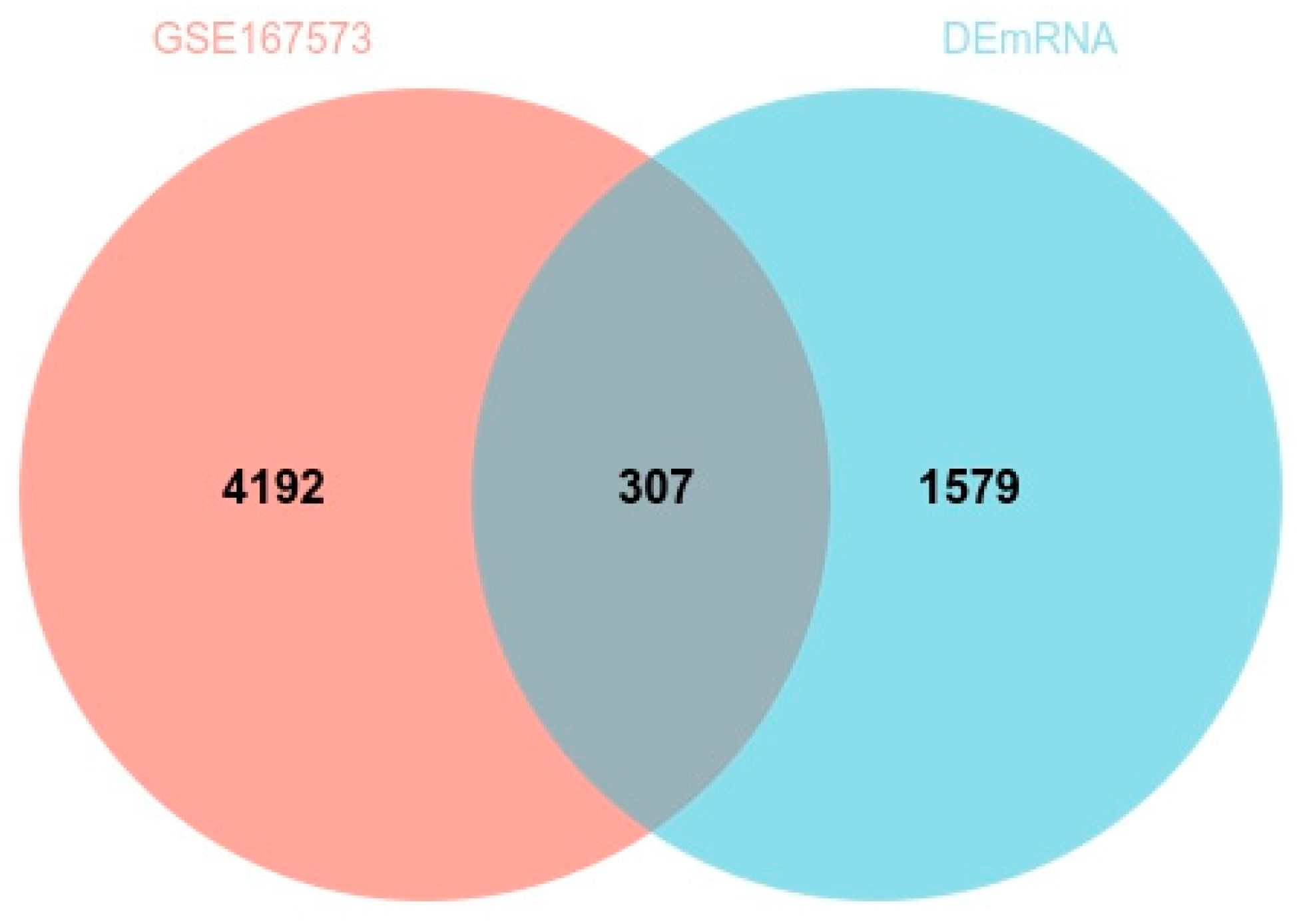
3.6. GEO Data Validation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ellis, C.L.; Eble, J.N.; Subhawong, A.P.; Martignoni, G.; Zhong, M.; Ladanyi, M.; Epstein, J.I.; Netto, G.J.; Argani, P. Clinical heterogeneity of Xp11 translocation renal cell carcinoma: Impact of fusion subtype, age, and stage. Mod. Pathol. 2014, 27, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Calió, A.; Grignon, D.J.; Stohr, B.A.; Williamson, S.R.; Eble, J.N.; Cheng, L. Renal cell carcinoma with TFE3 translocation and succinate dehydrogenase B mutation. Mod. Pathol. 2017, 30, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Kuthi, L.; Somorácz, Á.; Micsik, T.; Jenei, A.; Hajdu, A.; Sejben, I.; Imre, D.; Pósfai, B.; Kóczián, K.; Semjén, D.; et al. Clinicopathological Findings on 28 Cases with XP11.2 Renal Cell Carcinoma. Pathol. Oncol. Res. 2020, 26, 2123–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argani, P. Translocation carcinomas of the kidney. Genes Chromosomes Cancer 2022, 61, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Choo, M.S.; Jeong, C.W.; Song, C.; Jeon, H.G.; Seo, S.I.; Hong, S.K.; Byun, S.S.; Chung, J.S.; Hong, S.H.; Hwang, E.C.; et al. Clinicopathologic characteristics and prognosis of Xp11.2 translocation renal cell carcinoma: A multicenter, propensity score matching analysis. Clin. Genitourin. Cancer 2017, 15, e819–e825. [Google Scholar] [CrossRef]
- van der Beek, J.N.; Hol, J.A.; Coulomb-l’Hermine, A.; Graf, N.; van Tinteren, H.; Pritchard-Jones, K.; Houwing, M.E.; de Krijger, R.R.; Vujanic, G.M.; Dzhuma, K.; et al. Characteristics and outcome of pediatric renal cell carcinoma patients registered in the International Society of Pediatric Oncology (SIOP) 93-01, 2001 and UK-IMPORT database: A report of the SIOP-Renal Tumor Study Group. Int. J. Cancer 2021, 148, 2724–2735. [Google Scholar] [CrossRef]
- van der Beek, J.N.; Geller, J.I.; de Krijger, R.R.; Graf, N.; Pritchard-Jones, K.; Drost, J.; Verschuur, A.C.; Murphy, D.; Ray, S.; Spreafico, F.; et al. Characteristics and Outcome of Children with Renal Cell Carcinoma: A Narrative Review. Cancers 2020, 12, 1776. [Google Scholar] [CrossRef] [PubMed]
- Komai, Y.; Fujiwara, M.; Fujii, Y.; Mukai, H.; Yonese, J.; Kawakami, S.; Yamamoto, S.; Migita, T.; Ishikawa, Y.; Kurata, M.; et al. Adult Xp11 translocation renal cell carcinoma diagnosed by cytogenetics and immunohistochemistry. Clin. Cancer Res. 2009, 15, 1170–1176. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, J.S.; Malik, F.; Amin, M.B.; Argani, P.; Bahrami, A. MiT family translocation renal cell carcinomas: A 15th-anniversary update. Histol. Histopathol. 2020, 35, 125–136. [Google Scholar] [PubMed]
- Qi, X.; Zhang, D.H.; Wu, N.; Xiao, J.H.; Wang, X.; Ma, W. ceRNA in cancer: Possible functions and clinical implications. J. Med. Genet. 2015, 52, 710–718. [Google Scholar] [CrossRef]
- Yu, F.; Liu, J.B.; Wu, Z.J.; Xie, W.T.; Zhong, X.J.; Hou, L.K.; Wu, W.; Lu, H.M.; Jiang, X.H.; Jiang, J.J.; et al. Tumor suppressive microRNA-124a inhibits stemness and enhances gefitinib sensitivity of non-small cell lung cancer cells by targeting ubiquitin-specific protease 14. Cancer Lett. 2018, 427, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhu, D.; Li, H.; Li, H.; Feng, C.; Zhang, W. Characterization of circRNA-associated-ceRNA networks in a senescence-accelerated mouse prone 8 brains. Mol. Ther. 2017, 25, 2053–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, K.J.; Ma, Y.S.; Jiang, X.H.; Wu, T.M.; Wu, Z.J.; Li, Z.Z.; Wang, J.H.; Gao, Q.X.; Yi, B.; Shi, Y.; et al. Whole-Transcriptome Sequencing Identifies Key Differentially Expressed mRNAs, miRNAs, lncRNAs, and circRNAs Associated with XP11 TRANSLOCATION RCC. Mol. Ther. Nucleic Acids 2020, 21, 592–603. [Google Scholar] [CrossRef]
- Li, Q.; Dai, Y.; Wang, F.; Hou, S. Differentially expressed long non-coding RNAs and the prognostic potential in colorectal cancer. Neoplasma 2016, 63, 977–983. [Google Scholar] [CrossRef]
- Mishra, S.; Yadav, T.; Rani, V. Exploring miRNA-based approaches in cancer diagnostics and therapeutics. Crit. Rev. Oncol. Hematol. 2016, 98, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, F.; Ma, J.; Liu, Y.; Wang, X.; Wang, R.; Zhang, Y.; Zhang, W.; He, Q.; Song, D.; et al. Study on the Relationship between the miRNA-centered ceRNA Regulatory Network and Fatigue. J. Mol. Neurosci. 2018, 40, 3803–3811. [Google Scholar] [CrossRef]
- Saeidian, A.H.; Youssefian, L.; Vahidnezhad, H.; Uitto, J. Research Techniques Made Simple: Whole-Transcriptome Sequencing by RNA-Seq for Diagnosis of Monogenic Disorders. J. Investig. Dermatol. 2020, 140, 1117–1126.e1. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Liu, Y.; Peng, J.; Sun, T.; Li, N.; Zhang, L.; Ren, J.; Yuan, H.; Kan, S.; Pan, Q.; Li, X.; et al. Epithelial EZH2 serves as an epigenetic determinant in experimental colitis by inhibiting TNFα-mediated inflammation and apoptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E3796–E3805. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.M.; Yi, W.W.; Ma, Y.S.; Wu, W.; Yu, F.; Fan, H.W.; Lv, Z.W.; Yang, H.Q.; Chang, Z.Y.; Zhang, C.; et al. Prognostic implications of decreased microRNA-101-3p expression in patients with non-small cell lung cancer. Oncol. Lett. 2018, 16, 7048–7056. [Google Scholar] [CrossRef] [Green Version]
- Nayak, C.; Singh, S.K. Integrated Transcriptome Profiling Identifies Prognostic Hub Genes as Therapeutic Targets of Glioblastoma: Evidenced by Bioinformatics Analysis. ACS Omega 2022, 7, 22531–22550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Ma, Y.S.; Xia, Q.; Yu, F.; Lv, Z.W.; Jia, C.Y.; Jiang, X.X.; Zhang, L.; Shao, Y.C.; Xie, W.T.; et al. MicroRNA-mRNA integrated analysis based on a case of well-differentiated thyroid cancer with both metastasis and metastatic recurrence. Oncol. Rep. 2018, 40, 3803–3811. [Google Scholar] [CrossRef] [Green Version]
- Bandettini, W.P.; Kellman, P.; Mancini, C.; Booker, O.J.; Vasu, S.; Leung, S.W.; Wilson, J.R.; Shanbhag, S.M.; Chen, M.Y.; Arai, A.E. MultiContrast Delayed Enhancement (MCODE) improves detection of subendocardial myocardial infarction by late gadolinium enhancement cardiovascular magnetic resonance: A clinical validation study. J. Cardiovasc. Magn. Reson. 2012, 14, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Zhou, M.; Zou, L.; Miao, J.; Wang, Y.; Li, Y.; Lu, S.; Yu, J. Long non-coding RNA LOXL1-AS1 acts as a ceRNA for miR-324-3p to contribute to chol-angiocarcinoma progression via modulation of ATP-binding cassette transporter A1. Biochem. Biophys. Res. Commun. 2019, 513, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Cheng, X.; Gan, W.; Zhang, G.; Li, X.; Guo, H. Clinical characteristics of XP11.2 translocation/TFE3 gene fusion renal cell carcinoma: A systematic review and meta-analysis of observational studies. BMC Urol. 2016, 16, 40. [Google Scholar] [CrossRef] [Green Version]
- Malouf, G.G.; Camparo, P.; Oudard, S.; Schleiermacher, G.; Theodore, C.; Rustine, A.; Dutcher, J.; Billemont, B.; Rixe, O.; Bompas, E.; et al. Targeted agents in metastatic Xp11 translocation/TFE3 gene fusion renal cell carcinoma (RCC): A report from the Juvenile RCC Network. Ann. Oncol. 2010, 21, 1834–1838. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumor and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstråhle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef]
- Macrae, T.; Sargeant, T.; Lemieux, S.; Hébert, J.; Deneault, E.; Sauvageau, G. RNA-Seq reveals spliceosome and proteasome genes as most consistent transcripts in human cancer cells. PLoS ONE 2013, 8, e72884. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Meng, F.; Jiang, R. Neutrophil-to-Lymphocyte Ratio as a Prognostic Biomarker for Patients with Metastatic Renal Cell Carcinoma Treated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 746976. [Google Scholar] [CrossRef]
- Leitch, E.F.; Chakrabarti, M.; Crozier, J.E.M.; McKee, R.F.; Anderson, J.H.; Horgan, P.G.; McMillan, D.C. Comparison of the Prognostic Value of Selected Markers of the Systemic Inflammatory Response in Patients with Colorectal Cancer. Br. J. Cancer 2007, 97, 1266–1270. [Google Scholar] [CrossRef]
- Guthrie, G.J.; Charles, K.A.; Roxburgh, C.S.; Horgan, P.G.; McMillan, D.C.; Clarke, S.J. The Systemic Inflammation-Based Neutrophil-Lymphocyte Ratio: Experience in Patients with Cancer. Crit. Rev. Oncol./Hematol. 2013, 88, 218–230. [Google Scholar] [CrossRef]
- Nunno, V.D.; Mollica, V.; Gatto, L.; Santoni, M.; Cosmai, L.; Porta, C.; Massari, F. Prognostic Impact of Neutrophil-to-Lymphocyte Ratio in Renal Cell Carcinoma: A Systematic Review and Meta-Analysis. Immunotherapy 2019, 11, 631–643. [Google Scholar] [CrossRef]
- Su, S.; Liu, L.; Li, C.; Zhang, J.; Li, S. Prognostic Role of Pretreatment Derived Neutrophil to Lymphocyte Ratio in Urological Cancers: A Systematic Review and Meta-Analysis. Int. J. Surg. 2019, 72, 146–153. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Perera, R.M.; Di Malta, C.; Ballabio, A. MiT/TFE family of transcription factors, lysosomes, and cancer. Annu. Rev. Cancer Biol. 2019, 3, 203–222. [Google Scholar] [CrossRef]
- Bian, B.; Mongrain, S.; Cagnol, S.; Langlois, M.J.; Boulanger, J.; Bernatchez, G.; Carrier, J.C.; Boudreau, F.; Rivard, N. Cathepsin B promotes colorectal tumorigenesis, cell invasion, and metastasis. Mol. Carcinog. 2016, 55, 671–687. [Google Scholar] [CrossRef]
- Brisson, L.; Bański, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.J.; Van Hée, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Jia, E.; Ren, N.; Xin, H. Identification of prognostic miRNA biomarkers for esophageal cancer based on The Cancer Genome Atlas and Gene Expression Omnibus. Medicine 2021, 100, e24832. [Google Scholar] [CrossRef]
- Gao, X.; Liu, H.; Wang, R.; Huang, M.; Wu, Q.; Wang, Y.; Zhang, W.; Liu, Y. Hsa-let-7d-5p Promotes Gastric Cancer Progression by Targeting PRDM5. J. Oncol. 2022, 2022, 2700651. [Google Scholar] [CrossRef]
- Li, K.; Yao, T.; Zhang, Y.; Li, W.; Wang, Z. NEAT1 as a competing endogenous RNA in tumorigenesis of various cancers: Role, mechanism, and therapeutic potential. Int. J. Biol. Sci. 2021, 17, 3428–3440. [Google Scholar] [CrossRef]
- Klec, C.; Prinz, F.; Pichler, M. Involvement of the long noncoding RNA NEAT1 in carcinogenesis. Mol. Oncol. 2019, 13, 46–60. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wang, Y.; Liu, Z.; Zhang, Z.; Xiong, L.; Wen, Y. Recent advances of NEAT1-miRNA interactions in cancer. Acta Biochim. Biophys. Sin. 2022, 54, 153–162. [Google Scholar] [CrossRef]
- Ning, L.; Li, Z.; Wei, D.; Chen, H.; Yang, C. LncRNA, NEAT1 is a prognosis biomarker and regulates cancer progression via epithelial-mesenchymal transition in clear cell renal cell carcinoma. Cancer Biomark. 2017, 19, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liao, X.; Cheng, J.; Su, G.; Yuan, F.; Zhang, Z.; Wu, J.; Mei, H.; Tan, W. Targeted Methylation of the LncRNA NEAT1 Suppresses Malignancy of Renal Cell Carcinoma. Front. Cell Dev. Biol. 2021, 9, 777349. [Google Scholar] [CrossRef]
- Moreno-García, L.; López-Royo, T.; Calvo, A.C.; Toivonen, J.M.; de la Torre, M.; Moreno-Martínez, L.; Molina, N.; Aparicio, P.; Zaragoza, P.; Manzano, R.; et al. Competing Endogenous RNA Networks as Biomarkers in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9582. [Google Scholar] [CrossRef]
- Niu, Z.S.; Wang, W.H.; Dong, X.N. Role of long noncoding RNA-mediated competing endogenous RNA regulatory network in hepatocellular carcinoma. World J. Gastroenterol. 2020, 26, 4240–4260. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, C.; Wei, C.; Hou, Y.; Xiong, M.; Ni, D.; Huang, Y.; Wang, M.; Yang, X.; Chen, K.; Chen, Z. Identification of Key Differentially Expressed mRNAs, miRNAs, lncRNAs, and circRNAs for Xp11 Translocation Renal Cell Carcinoma (RCC) Based on Whole-Transcriptome Sequencing. Genes 2023, 14, 723. https://doi.org/10.3390/genes14030723
Deng C, Wei C, Hou Y, Xiong M, Ni D, Huang Y, Wang M, Yang X, Chen K, Chen Z. Identification of Key Differentially Expressed mRNAs, miRNAs, lncRNAs, and circRNAs for Xp11 Translocation Renal Cell Carcinoma (RCC) Based on Whole-Transcriptome Sequencing. Genes. 2023; 14(3):723. https://doi.org/10.3390/genes14030723
Chicago/Turabian StyleDeng, Changqi, Chengcheng Wei, Yaxin Hou, Ming Xiong, Dong Ni, Yu Huang, Miao Wang, Xiong Yang, Ke Chen, and Zhaohui Chen. 2023. "Identification of Key Differentially Expressed mRNAs, miRNAs, lncRNAs, and circRNAs for Xp11 Translocation Renal Cell Carcinoma (RCC) Based on Whole-Transcriptome Sequencing" Genes 14, no. 3: 723. https://doi.org/10.3390/genes14030723